Introduction
At Neph Cure Inc., we are dedicated to advancing the understanding and treatment of rare kidney diseases. This blog focuses on two critical topics: IgAN diagnosis and Infantile Nephrotic Syndrome causes. By shedding light on these conditions, we aim to provide valuable information for patients, caregivers, and healthcare professionals.
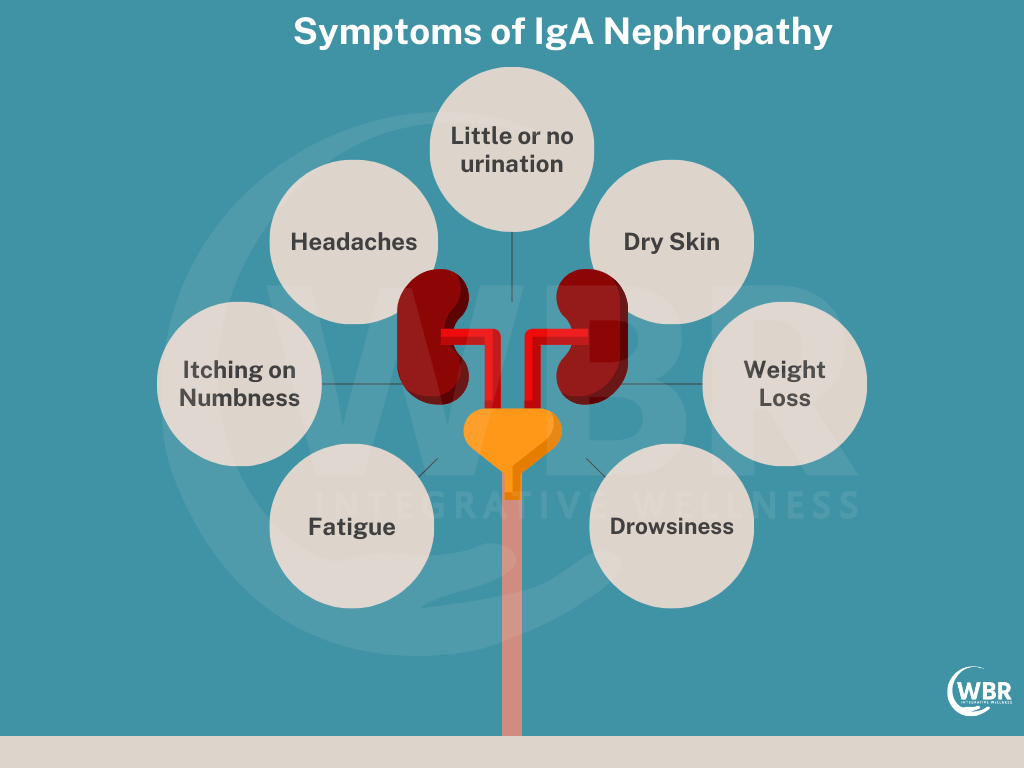
IgAN Diagnosis
IgA Nephropathy (IgAN), also known as Berger’s disease, is a kidney disorder caused by the buildup of the antibody immunoglobulin A (IgA) in the glomeruli. Accurate diagnosis is crucial for effective management and treatment.
1. Urine Tests: The first step in diagnosing IgAN often involves urine tests. These tests can detect the presence of blood (hematuria) and protein (proteinuria) in the urine, which are common indicators of kidney damage.
2. Blood Tests: Blood tests are used to assess kidney function by measuring levels of creatinine and blood urea nitrogen (BUN). Elevated levels of these substances can indicate impaired kidney function. Additionally, blood tests can measure the levels of IgA antibodies.
3. Kidney Biopsy: A kidney biopsy is the definitive test for diagnosing IgAN. During this procedure, a small sample of kidney tissue is removed and examined under a microscope. The presence of IgA deposits in the glomeruli confirms the diagnosis of IgAN.
4. Imaging Tests: Imaging tests such as ultrasound or CT scans may be used to evaluate the size and structure of the kidneys. These tests can help rule out other conditions that may cause similar symptoms.
5. Genetic Testing: In some cases, genetic testing may be recommended, especially if there is a family history of IgAN. Genetic testing can identify mutations that may predispose individuals to the disease.
Infantile Nephrotic Syndrome Causes
Infantile Nephrotic Syndrome is a rare kidney disorder that typically presents within the first three months of life. Understanding the causes of this condition is essential for early diagnosis and effective management.
1. Genetic Mutations: Genetic mutations are the primary cause of Infantile Nephrotic Syndrome. Mutations in genes such as NPHS1 (nephrin), NPHS2 (podocin), and WT1 (Wilms’ tumor 1) are commonly associated with the condition. These genes play crucial roles in the structure and function of the glomerular filtration barrier. Mutations disrupt this barrier, leading to excessive protein loss in the urine.
2. Inherited Patterns: Infantile Nephrotic Syndrome can be inherited in an autosomal recessive manner, meaning both parents must carry a copy of the mutated gene for the child to be affected. Genetic counseling and testing are essential for families with a history of the condition to understand their risk and make informed decisions.
3. Prenatal Factors: In some cases, Infantile Nephrotic Syndrome may be detected prenatally through ultrasound findings of fetal edema or a large placenta. Early diagnosis allows for prompt intervention and management after birth.
4. Environmental Triggers: While genetic mutations are the primary cause, environmental factors may also play a role in the severity and progression of Infantile Nephrotic Syndrome. Infections and other stressors can exacerbate the condition, highlighting the importance of comprehensive care and monitoring.
5. Secondary Causes: In rare instances, Infantile Nephrotic Syndrome may be secondary to other conditions such as congenital infections or metabolic disorders. Identifying and treating the underlying cause is crucial for effective management.
Conclusion
Neph Cure Inc. is committed to raising awareness and providing support for individuals affected by IgA Nephropathy and Infantile Nephrotic Syndrome. By understanding the diagnosis process for IgAN and the causes of Infantile Nephrotic Syndrome, we can work towards better diagnosis, treatment, and ultimately, improved outcomes for patients and their families. Stay informed and join us in our mission to combat rare kidney diseases.